



.png)

.png)
The Kitchen We Didn’t Design: Lived Experience as Advocacy

March 25, 2026

For the first time in the history of ALS, the drug development pipeline is full of promising therapies. In this volume of Synapticure on Science, we share information about some of the investigational therapies we get asked about most often for sporadic ALS:
The above list includes interventional drug trials for sporadic ALS. Later this year, a future edition of Synapticure on Science wIll summarize observational ALS trials, as well as interventional drug trials for genetic forms of ALS.
For those trials currently enrolling, the anticipated completion dates range from April 2025 to July 2026. The quicker those trials are fully enrolled, the quicker the trial investigators can release those data, and potentially seek FDA approval if the trials meet their endpoints or have promising biomarker data. If you are interested in understanding trial qualifications and factors that might impact which clinical trial could be best for you and your family, we encourage you to read our inaugural edition of Synapticure on Science and contact your care coordinator to schedule an appointment.
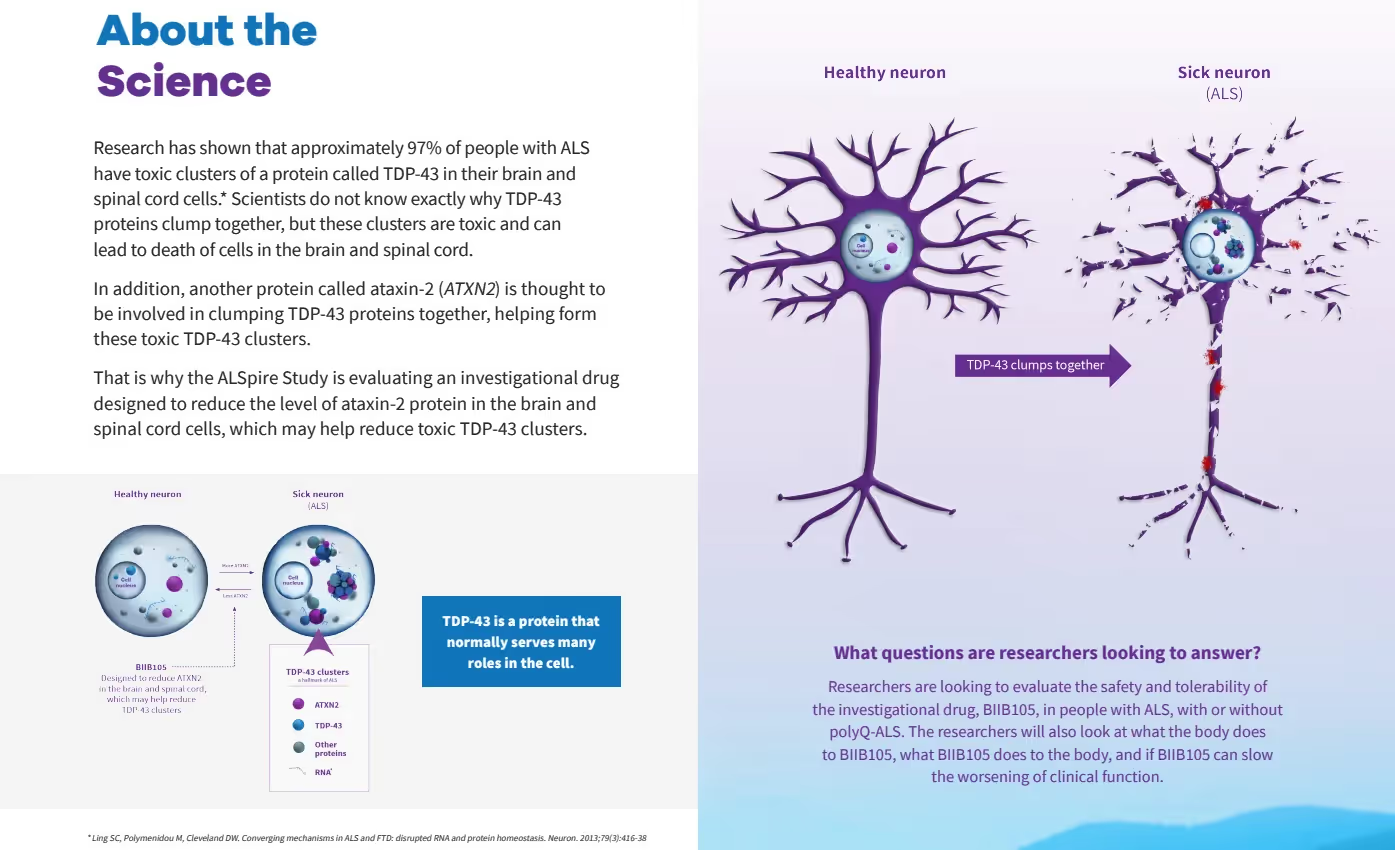
Despite the vast heterogeneity of sporadic and familial ALS, one thing is nearly universally true. Approximately 97% of people with ALS have TDP-43 aggregates in their motor neurons, the nerve cells that control voluntary movement and are progressively damaged in ALS. Additionally, nearly 50% of people with frontotemporal dementia (FTD) also have TDP-43 aggregation.
Many of our patients ask: ‘What is TDP-43 and what does it do in ALS?’
TDP-43 is a protein. Normally TDP-43 sits in the nucleus of the cell. But in ALS and FTD, it is “mislocalized,” moving from the nucleus to the cytoplasm in neurons and glial cells in the central nervous system. In 2008, researchers concluded that TDP-43 disruption plays a role in the pathology of people with both familial and sporadic forms of ALS. Since then, the pre-clinical work studying TDP-43 and ALS has exploded; over the last five years, there has been over 200 peer-reviewed publications, annually, on PubMed.gov related to TDP-43 and ALS.
Numerous pre-clinical studies in the lab and in animal models showed that both TDP-43 mutations and TDP-43 overexpression are associated with ALS.
Because TDP-43 pathology occurs in nearly all types of ALS, it has become an important therapeutic target in the drug development pipeline. Indeed, demonstrating the importance of this science, in September, top ALS researchers from around the world are meeting for the First Annual Bi-ennial Conference on TDP-43 Function and Dysfunction in Disease. They will be discussing: ALS and FTD; events driving TDP-43 mislocalization and aggregation; impact of TDP-43 pathology on neuronal integrity; and new therapeutic directions. Some of those therapeutic options are exemplified in the following clinical trials.
Biogen is testing an antisense oligonucleotide (ASO) called BIIB-105 that is delivered via intrathecal injection. This ASO was initially developed by Ionis and called ION-541.
Ionis’ scientists specialize in ASO technology. They innovated and developed the recently approved ASO, Tofersen, targeting the SOD1 mutation, as well as the drug called Spinraza for children with Spinal Muscular Atrophy. Now they have used that same technology to target the Ataxin-2 (ATXN2) protein in ALS. The reduction of ATXN2 has been shown to decrease aggregation of TDP-43.
The BIIB-105 study is called the “ALSpire” study (NCT04494256 on clinicaltrials.gov). Its anticipated completion date is July 15, 2026. This image is an excerpt from Biogen’s brochure illustrating the science underlying the ALSpire study.
The ALSpire study consists of two parts:
ALSpire is a dose-escalating study investigating BIIB-105’s safety and tolerability in about 108 ALS patients with or without polyQ repeats in the ATXN2 gene.1 The study is recruiting 48 people with sporadic ALS.
The objectives of the study are to evaluate:
While ALSpire is not an efficacy trial, one of the stated objectives is to assess whether BIIB-105 can slow the loss of function.
Synapticure’s neurologists can discuss the risk-benefits of participating in a Phase 1 trial as well as your own risk tolerance and priorities so you and your family can make the best decision for you. One of the things we will discuss with you is the evidence developed in the preclinical studies and animal models.
For example, below is an image from the ALSpire presentation on January 12, 2023. Of interest are the results of the animal studies where ASOs that targeted ATXN2 protein levels caused “significant improvement in both survival and walking.”
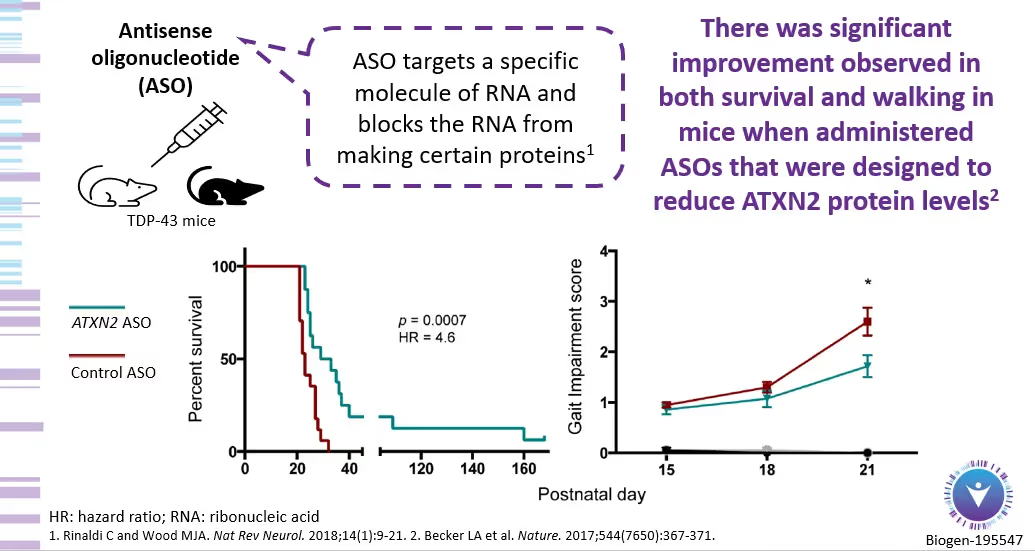
As with all Phase 1 studies, the downside is that there are no safety data yet for the therapy. But, with a two-year OLE – plus a possible six months of dosing in part 1 of the trial – the possible upside is IF the therapy shows efficacy, trial participants could potentially receive the therapy years before Biogen could ever apply for approval. This is the benefit that many saw when they were early participants in the Tofersen ASO trial targeting the SOD1 mutation. This does not mean BIIB-105 will have the same impact, but this is part of the informed risk-benefit dialogue our clinical team will discuss with you, along with a detailed discussion about the science underlying BIIB-105.
The ALSpire BIIB-105 study will require 50 visits total: 19 clinic visits in Part 1 and 31 clinic visits in Part 2. Thus, finding a convenient location is important. If you are interested in the BIIB-105 trial, there are trial sites located in 12 states across the US, plus one in Quebec and some in the EU. Biogen does offer some travel assistance.
The trial sites and Principal Investigators (PIs) in the US are:
The Healey Platform Trial is enrolling two regimens right now. Both regimens are small molecule drugs you take orally. Each will enroll approximately 240 people each, and have a 3:1 placebo ratio. The patient-centric trial also has crossover design in which all people will receive the treatment during an Open Label Extension after the end of the 24-week double-blinded trial. Both drugs aim to slow ALS progression by targeting harmful buildup of protein TDP-43 via the integrated stress response.
Both regimens are collecting biomarker data from blood and urine samples. While it is voluntary, both are also collecting samples of cerebral spinal fluid (CSF) via a lumbar puncture at the beginning and end of the trial. Collection of CSF is important to advance the science of ALS. But it’s especially critical to collect objective data that shows if an investigational therapy is having an intended effect on biological targets. This objective biomarker data can hopefully expedite eventual approval by the FDA.
The time to qualify for the Healey Platform trial is 36 months from symptom onset. The Master Protocol (NCT04297683) outlines other qualification criteria, including but not limited to: slow vital capacity ≥50%; you must be able to swallow pills or liquids; you cannot have active cancer or a history of cancer in the last 3 years; previous use of any gene therapy disqualifies you; and you cannot be taking any other product being studied in clinical trials.
The patient-centric Healey trial also has dozens of sites across the US. You need not be a US citizen to participate, but you must be “geographically accessible” to a trial site. Check the list for each regimen as not all sites are enrolling both regimens yet.
Healey is now enrolling the trial for ABBV-CLS-7262, developed by Calico Life Sciences in collaboration with large biopharma company, AbbVie. (NCT05740813 on ClinicalTrials.gov).
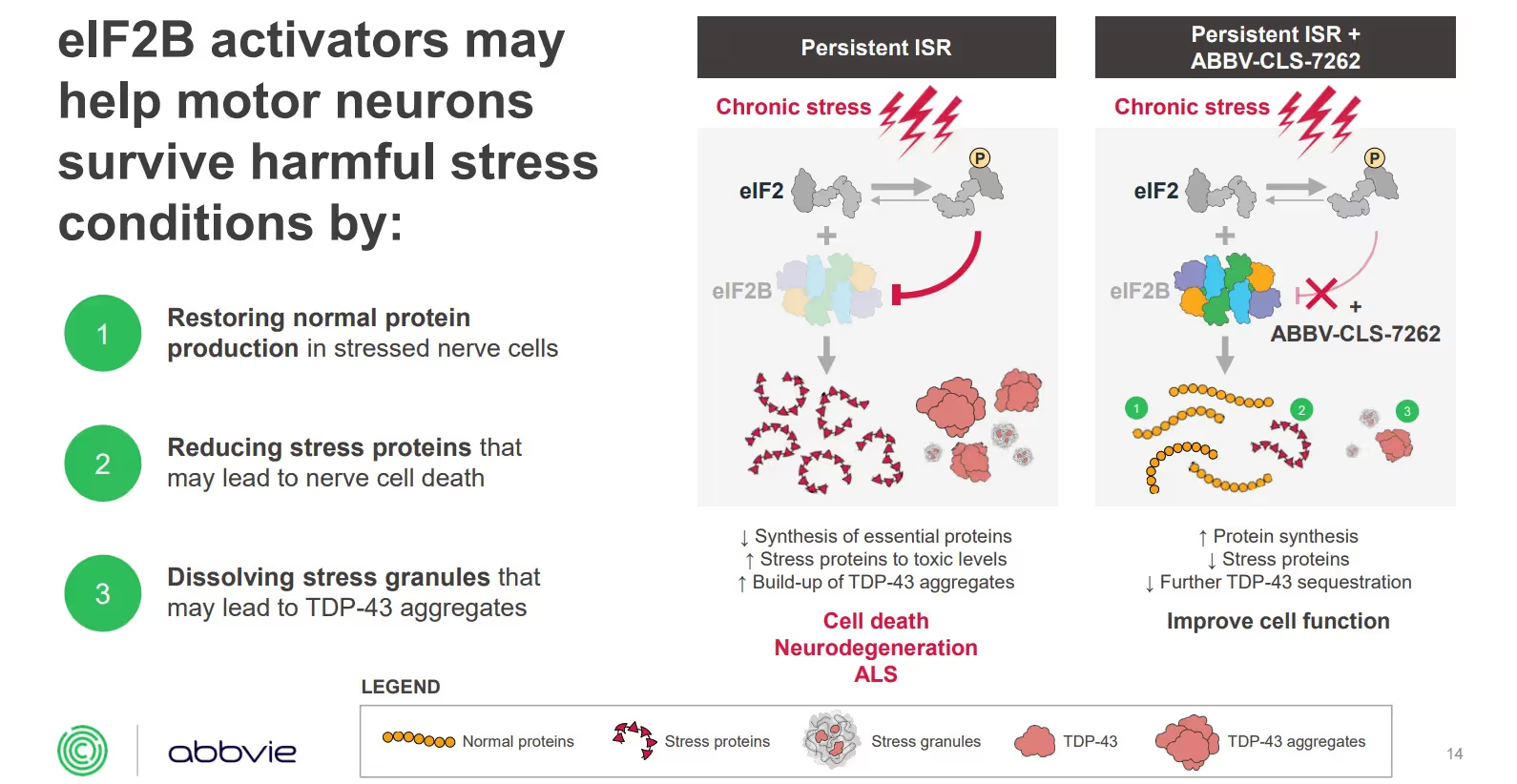
This investigational small molecule drug aims to activate eIF2B, a key regulator of the integrated stress response (ISR). During stress, toxic exposures and disease, the activity of eIF2B is inhibited. By activating elF2B, it may help motor neurons survive harmful stress conditions by: (a) restoring normal protein levels in stressed nerve cells; (b) reducing stress proteins that may lead to cell death; and (c) dissolving stress granules that may lead to TDP-43 aggregates. TDP-43 containing stress granules are thought to lead to TDP-43 inclusions, a hallmark of ALS pathology.
The trial’s primary endpoint is change in disease progression as measured by the ALSFRS-R score. Its secondary endpoints measure SVC and muscle strength.To learn more, you can watch the detailed Healey mechanism of action webinar. The following graphic is an excerpt from the webinar explaining the proposed mechanism of action:
The earlier Phase 1 study enrolled 31 people. (NCT04948645 on ClinicalTrials.gov). This trial is a two-part study that will last up to three years (156 weeks). Part 1 was a four-week, randomized, double-blind, placebo-controlled study. Part 2 is a 152-week Open Label Extension, during which all people participating in the small trial will receive ABBV-CLS-7262.
The anticipated completion date of the earlier Phase 1 study and OLE is 2025. The estimated completion date of the currently enrolling Phase 2/3 trial is April 2025.
Healey is now enrolling the trial of DNL343, developed by biopharma company Denali Therapeutics (NCT05842941 on ClinicalTrials.gov). On July 20th, Danna Jennings of Denali Therapeutics presented a mechanism of action webinar, along with lead investigator Dr. Merit Cudkowicz. You can watch that detailed webinar here.
The trial’s primary endpoint is change in disease progression as measured by the ALSFRS-R score. Its secondary endpoints measure change from baseline ALSFRS-R, SVC, muscle strength, and survival.
This graphic is from the corporate presentation showing DNL343’s impact on neurons and iPSC-derived neurons:
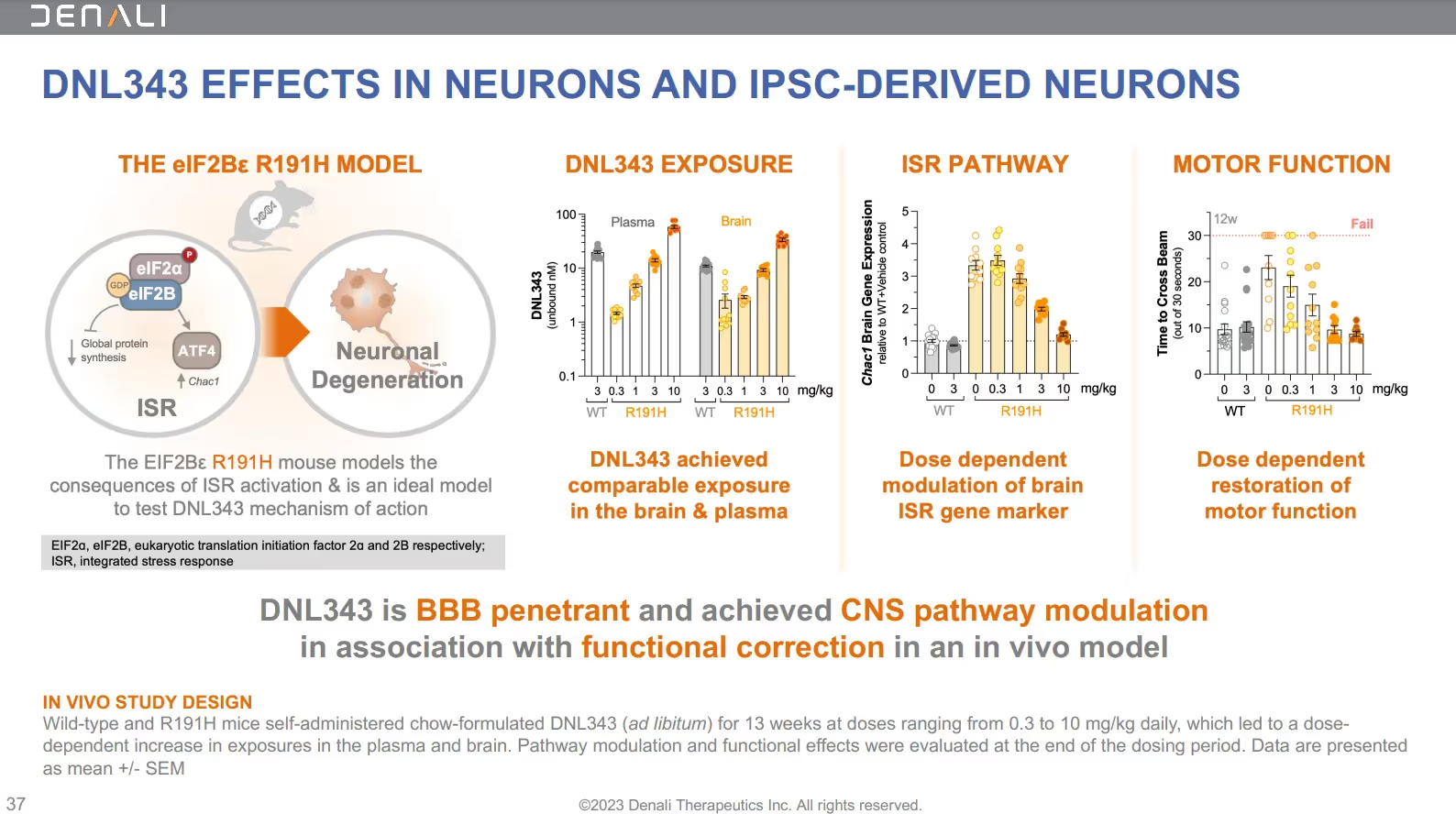
The earlier Phase 1b study of DNL343 enrolled 29 people in the randomized, placebo-controlled, double-blind study. This trial lasted 28 days – followed by an ongoing 18-month open-label extension (OLE), designed to evaluate the safety, pharmacokinetics, and pharmacodynamics of DNL343.
The results of the Phase 1b safety study and OLE have not yet been published. (NCT05006352 on ClinicalTrials.gov). However, Denali did present the preliminary Phase 1 data in a poster presentation at the American Academy of Neurology conference in April 2023:
The anticipated completion date of the earlier Phase 1b study and OLE of DNL343 is December 2023. The estimated completion date of the currently enrolling Phase 2/3 trial is May 2025.
QurAlis is a new biotech company with promising clinical and pre-clinical work in the pipeline for ALS therapies. Currently, its investigational drug, QRL-201, is in clinical trials in Canada (NCT05633459 on ClinicalTrials.gov). Just like the recently approved drug, Tofersen, QRL-201 uses ASO technology delivered via intrathecal injection.
QRL-201 is an investigational therapy that aims to restore STATHMIN-2 (STMN2) expression in people with ALS. Importantly, STMN2 expression is significantly decreased in nearly all ALS patients. STMN2 is the gene most significantly regulated by TDP-43 , and “the most consistently decreased gene over all sporadic ALS patient data sets.” Highly expressed in human motor neurons, STMN2 is an important protein for neural repair and axonal stability. In animal models, STMN2 deletion caused axonal degeneration, which leads to paralysis in ALS patients.
QRL-201 aims to rescue STMN2 loss of function. Recently, QurAlis’ Chief Medical Officer, Dr. Angela Genge, spoke to Neurology Live to explain QRL-201’s proposed mechanism of action and how it could “completely change” the current therapeutic approach for people who develop ALS related to TDP-43 pathology:
One of the first proteins to be knocked down when patients develop ALS with TDP-43 pathology is Stathmin…. This ASO, that is called QRL-201, by restoring Stathmin could stabilize the loss of motor neurons entirely, by returning to normal levels a protein that we believe is fundamental to the loss of motor neurons. We believe that if this is as effective as we see in the cell model, we can actually cause a stabilization of this disease….
QurAlis has announced that the first patients have been dosed with QRL-201 in its Phase 1 trial, with trial sites in Calgary and Quebec, Canada. It also has received regulatory clearance to conduct QRL-201 clinical trials in the UK and EU. The trial has a target enrollment of 62 people with ALS. The anticipated completion date of the Phase 1 trial is May 2025.
Pridopidine is being tested in the Healey Platform trial (NCT04615923). In this detailed mechanism of action webinar, you can learn more about the Pridopidine trial from Prilenia’s Dr. Michael Hayden, along with two of the trial investigators, Dr. Bjorn Oskarsson of Mayo Clinic, and Dr. Jeremy Shefner of Barrow Neurological Institute.
Prilenia’s Pridopidine is an investigational drug called a Sigma-1 Receptor (S1R) agonist. S1R is a protein that is highly expressed in the brain and spinal cord, where it regulates various processes that are often impaired in people with ALS. Although Pridopidine is being developed for sporadic ALS, it’s notable that a mutation in the gene that encodes S1R is also a cause of inherited forms of ALS. There is a gene-dose relationship: when there’s a complete loss of S1R, it presents as juvenile ALS. When there is a partial loss of S1R, people get adult-onset ALS.
In this short video, you can see how Pridopidine’s mechanism of action targets multiple cellular pathways essential to neuronal function and survival in sporadic ALS as well. Importantly, imaging studies confirm that Pridopidine passes the blood brain barrier and enters the brain and spinal cord where it activates S1R and:
Earlier this year, Prilenia released the topline results from its Phase 2 study of Pridopidine. While the trial didn’t meet its endpoints over the short six-month timeframe, there were some “consistent, positive trends” in motor function, in speech & bulbar function, and in disease-related biomarker neurofilament light (NfL).
In both pre-specified and post hoc analyses, fast progressors who were earlier in progression showed benefit on the ALSFRS scale. From many other ALS trials, the research community knows that it’s easier to detect a signal in fast progressors in short six-month trials so this outcome is not surprising. In this Phase 2/3 Pridopidine trial, fast progressors who were less than 18 months from symptom onset had “substantially less decline” with a 5+ point slower progression on Pridopidine than placebo.2
Importantly, the trial detected promising trends in the neurodegeneration biomarker, neurofilament light (NfL)—which was recently used to support approval of Tofersen.3 Neurofilament Light is a biomarker of ongoing neuronal injury that is increased in many neurodegenerative diseases—but it is most markedly increased in ALS. In post hoc analyses, once again, the recently diagnosed, fast progressors showed a biologically meaningful distinction. Pridopidine demonstrated an average of 40% reduction in NfL compared to placebo.4
Just as with the ALSFRS-R data regarding functional outcomes in domains of fine and gross motor skills, greater improvements in quantitative speech measures were observed among trial participants within 18 months of onset who also met criteria for definite ALS.
The press release from the Healey Center at MassGen stated that “pre-specified analyses of speech production demonstrated “substantial improvements” in participants taking Pridopidine compared to those taking placebo. Those improvements were observed in speaking rate, articulation rate, sustained phonation and articulatory precision.
For context, Synapticure’s speech-language pathologist (SLP), Kylee Haller, offered the following insight into what the normal baseline assessments are for some of these speech measures:
Pridopidine’s impact on speech mirrors what the Prilenia team anticipated might happen when designing the trial. In the mechanism of action webinar before the trial started, CEO and neurologist Dr. Michael Hayden was asked about why the trial design, uniquely, included several secondary endpoints that measured impact on bulbar function and speech. He responded by comparing off-label therapy, Nuedexta, with their investigational therapy Pridopidine:
“There is one other drug that’s been used in ALS that had significant impact—but it’s not a very selective Sigma-1, and as we explained before, selectivity is important… Nuedexta targets Sigma-1, but it targets a lot of other targets as well. But what we did see in some of the early trials, and Merit and others were involved in them, is that you saw some improvement in speech and swallowing, as measured by the bulbar scale. So as part of our secondary [endpoints], we put that higher because there was some evidence that Sigma-1 may be, actually, one of the earliest signs that you might manifest and you might be able to detect. But those trials were short-term … and we asked ourselves the question as whether Sigma-1 stimulation may manifest very early by changes in speech and swallowing, and that’s why it’s higher up in the secondary outcome measures.”
Based on this potential mechanism of action on bulbar and speech function, the trial collected and analyzed speech production using quantitative voice characteristics derived from data collected with a smartphone application from Aural Analytics. In fact, Pridopidine was the first ALS trial to show positive effects on digital measures of speech. Those secondary endpoints produced probative data that Pridopidine works on speech and bulbar function. Commenting on the importance of these findings, the Healey Center’s Dr. Merit Cudkowicz, said:
“In particular, the impact of pridopidine on speech measures was notable, likely due to its S1R mechanism of action. Speech is a highly clinically relevant endpoint in ALS studies, and more than 80% of ALS patients become speech impaired, which significantly impacts their quality of life.”
The Healey Pridopidine trial enrolled 162 participants. The Phase 2/3 results also provided additional safety data and validated prior comprehensive safety data in Huntington’s Disease.
In sum, there are a variety of very promising data from Prilenia’s Pridopidine trial. The ALS community is eagerly awaiting the results and release of the data from the longer Open Label Extension, and we’ll bring you that information in our Synapticure Synopsis as soon as it’s available.
Additionally, if you or your loved one did not meet the inclusion criteria for the Pridopidine trial, Prilenia does have a small expanded access program at a few trial sites. You can discuss this with your care team at Synapticure to see if this is an option for you.
To schedule an appointment with our Speech & Language Therapist, Care Coordinators or Neurologists, you can call (855) 255-5917 or click here.
In hopeful news, pharmaceutical company, CLENE, announced the publication of its data from the Australian Phase 2 study and Open Label Extension of its investigative ALS therapy, CNM-Au8 (aka gold nanocrystals). One of the trial investigators, Professor Steve Vucic of the University of Sydney said the treatment demonstrated:
The mechanism of action for CNM-Au8 is targeting energy metabolism. As our brains age, the ability to produce energy declines. Clene hypothesizes that these energy impairments both predispose people to neurodegenerative diseases and then continue to drive disease progression. By targeting energy metabolism, Clene believes CNM-Au8 may promote neuronal health. Clene hypothesizes that CNM-Au8 works in multiple ways: by increasing NAD, ATP and protein homeostasis, while decreasing oxidative stress.
CNM-Au8 is the same therapy being tested as Regimen C in the Healey Platform Trial here in the US (NCT04414345 on ClinicalTrials.gov). The Phase 2/3 Healey trial of CNM-Au8 is completed. There is an ongoing Expanded Access program at over a dozen sites across the US.
To learn more about the proposed mechanism of action of this therapy, you can watch the Healey webinar. In the 24-week Phase 2 Healey trial, CNM-Au8 did not meet its primary or secondary endpoints. However, in both the Australian Rescue-ALS study and in the Healey ALS study, they observed a survival benefit with the longer dosing period in OLE. As such, Clene said they plan to meet with the FDA in the third quarter of 2023 to identify next steps in the development pathway for CNM-Au8 for ALS. Additionally, in its investor presentation, Clene states it hopes to advance CNM-Au8 to a longer Phase 3 study called RESTORE-ALS. Once they report an update, we will bring that to you in our Synapticure Synopsis.
In exciting recent news, on July 3rd, the FDA gave Akava Therapeutics the okay to proceed with its first-in-human testing of its AKV9 (previously known as NU-9). This drug, AKV9, targets diseased upper motor neurons.
To receive an ALS diagnosis, you must have degeneration of both upper (cortical) and lower (spinal) motor neurons. They work together so your brain can communicate with your muscles. Upper motor neurons start in the brain and connect with and send messages to the lower motor neurons in the spinal cord, which in turn innervate and send messages to the voluntary muscles telling them to move. Yet, until now, no therapies have focused exclusively on rescuing upper motor neurons (UMN). AKV9 is a protein aggregation inhibitor that is the first compound targeting the health of UMNs that degenerate and die in ALS and primary lateral sclerosis (PLS).
Akava Therapeutics was started by Northwestern University researcher and chemist, Dr. Richard Silverman, who was responsible for the discovery of the blockbuster drug Lyrica that Pfizer brought to market. The name, Akava, means “inhibition” in Yiddish. As it pertains to ALS, the focus of the company’s drug development program is inhibition of protein aggregation.
Protein aggregation is the hallmark of not just ALS, but all neurodegenerative diseases. In the creation of AKV9, Dr. Silverman’s lab screened over 50,000 compounds to find those that inhibited protein aggregation. Ultimately, that led to narrowing the study to three optimized molecules, and they asked: which were the most stable, which had the least side effects and which best crossed the blood-brain barrier? The answer was NU-9, now known as AKV9.
Dr. Silverman has worked hand-in-hand with Dr. Hande Ozdinler, who developed different cellular and mouse models to test drugs’ efficacy on UMNs. First she developed a line where UMN were fluorescent, making them easier to study. Then she crossed that line with ALS mouse models to test if the therapy worked – not just in SOD1 models, which had led to dozens of failures in ALS drugs or sporadic ALS. Dr. Ozdinler also developed mouse models testing the drug on TDP-43, which is associated with most people with ALS.
Because ALS is so diverse in cellular pathology, Dr. Ozdinler told Endpoints News:
“ALS is a very heterogeneous disease. There are many underlying causes that lead to ALS, and one of the challenges for drug discovery was to find one compound that hits multiple birds all at the same time because there are so many different avenues to improve.”
You can watch this short three-minute video from Nature that illustrates some of these heterogeneous disease processes in people with ALS.
AKV9 has five different mechanisms of action—all focused on UMNs.
Northwestern media reported the promising effects of NU-9 (now known as AKV9):
After administering NU-9, both the mitochondria (the cell’s energy producer) and the endoplasmic reticulum (the cell’s protein producer) began to regain their health and integrity resulting in improved neuron health. The upper motor neurons were more intact, their cell bodies were larger and the dendrites were not riddled with holes. They stopped degenerating so much that the diseased neurons became similar to healthy control neurons after 60 days of NU-9 treatment.
Akava reported that it had filed its Investigational New Drug application with the FDA in May of 2023 and it was granted on July 3rd. First the company will start a Phase 1a study in healthy volunteers, perhaps before the end of 2023. Then, if the Phase 1a safety study shows no toxicity in people without ALS, it can then start a Phase 1b/2a trial in which the drug could be given to people with ALS hopefully in 2024.
To stay updated about when the trials begin enrolling, register for our email updates about ALS and other neurodegenerative diseases in our Synapticure Synopsis.
Since 2019, a much anticipated therapy is based on the science of regulatory T cells commonly referred to as “Tregs.” In ALS, this Treg research was initially led by Dr. Stanley Appel of Houston Methodist Hospital.
After decades of research related to the immune system, Dr. Appel’s team discovered that regulatory T cells were a key component accelerating ALS progression. These Tregs are immune cells that help protect the body from harmful inflammation that accelerates the progression of ALS. According to Dr. Appel:
“We found that many of our ALS patients not only had low levels of Tregs, but also that their Tregs were not functioning properly. We believed that improving the number and function of Tregs in these patients would affect how their disease progressed.”
In this video, Dr. Appel shared the results of the early Phase 1 study where they harvested the toxic Treg cells from people with ALS, expanded them until they became normal, then reinjected them back into the patients’ CSF via lumbar puncture. These three patients stopped progressing. But when the researchers stopped the infusions, the patients deteriorated. The researchers had a second opportunity to go back and give them Tregs, and once again they stopped ALS progression while receiving the therapy.
Recently, Dr. Appel joined a new company, COYA Therapeutics, to lead its Scientific Advisory Board and its development of therapies using Treg technology.
Now COYA has built upon Dr. Appel’s research and conducted a proof-of-concept (POC) study with its therapy called COYA-302. In this study, COYA’s team used subcutaneous injection of interleukin-2 in a combination T-reg therapy that can be scalable for manufacturing and large studies. Although it was only a small open label study with four people, the POC early study of COYA 302 showed promising results.
Prior to receiving COYA 302, all four people with ALS had been experiencing a significant decline in function.
Obviously this small four-person trial is not statistically powered for conclusions about efficacy. But it was a promising proof of concept, as once again, the COYA Treg findings mirrored the earlier Phase 1 study. ALS loss of function was significantly slowed or halted – as demonstrated by changes in the ALSFRS-R scores:
Additionally at a cellular level, COYA 302:
According to its press release, COYA plans to present and/or publish its biomarker data in the second half of 2023 concurrently with preparing for a meeting with the FDA in the fall of 2023. If the FDA gives the okay, COYA hopes to start a large, double-blind placebo controlled Phase 2 trial in 2024.
Masitinib is an oral pill. It is approved in the US and the EU for the treatment of mast cell tumors in dogs. It is not approved for use in humans.
Currently, masitinib is the only tyrosine kinase inhibitor in late-stage development for ALS. Two of masitinib's cellular targets are mast cells and microglia, both of which play a key role in inflammation. As such, it is hypothesized to slow disease progression and ease ALS symptoms.
Masitinib was studied in a Phase 2/3 clinical trial (NCT02588677) that compared the efficacy and safety of masitinib in combination with riluzole (masitinib combo) versus placebo (riluzole alone). The 48-week trial enrolled 394 people and ended in 2018. The study results were published in 2021. At the higher dose, it showed:
However, there was not a survival benefit in the overall trial population. But when AB Science limited the analysis to only those people earlier in progression, it minimized the floor effect of the ALSFRS-R. By examining the trial data for only those who had at least two points on every ALSFRS-R question at baseline, researchers found significant survival benefits. In this subgroup, patients on the masitinib combo lived for an average of 69 months versus 44 months for those only on riluzole. This resulted in a 44% reduction in the risk of death and an average additional survival of 25 months.
AB Science has submitted its application for Conditional Approval to both the Canadian and EU regulatory authorities (the equivalent to the FDA). Without biomarkers to be used as surrogate endpoints, there is no similar expedited approval pathway in the US. There is, however, an ongoing phase 3 study that can be used as the confirmatory trial for the EU application as well as a trial to support an application for approval in the US.
The time to qualify for the trial is 24 months fromsymptom onset. The trial protocol (NCT03127267) outlines other qualification criteria, including but not limited to: FVC ≥60%; baseline ALSFRS-R score ≥26, and scores of at least 2 on each of the 12 questions. Plus, ALSFRS-R score progression from symptom onset to screening must be > 0.3 per month, and confirmed with an ALSFRS-R score progression of ≥ 1 point during the 12-week run-in period between screening and randomization.
The trial is predominantly being conducted in the EU, but there are a few sites in the US: Johns Hopkins (MD), University of Southern California, University of Kentucky, University of Virginia, University of Alabama-Birmingham, and Lahey Hospital (MA).
The projected enrollment in the 48-week trial is 495 people. The trial’s primary endpoint is change from baseline ALSFRS-R score. Its secondary endpoints measure FVC, muscle strength, quality of life, and progression-free survival. The original anticipated completion date was December 2023.
The FDA’s decision about NurOwn (aka Debamestrocel) is one of the most eagerly anticipated in the ALS community. NurOwn is a mesenchymal stem cell therapy, unique because it is treated with neurotrophic factors. Neurotrophic factors are biomolecules that support the growth, survival and differentiation of neurons. In test tubes and animal models, they are capable of regrowing damaged neurons.
To make NurOwn, each person’s stem cells are harvested via a bone marrow aspiration. After the stem cells are treated with NurOwn in a lab for approximately one month, they are then reinjected into the patient’s CSF via an intrathecal injection (aka lumbar puncture). You can learn more about NurOwn technology and its proposed mechanism of action on pathways of neuroinflammation and neuroprotection by watching this short video.
Recently, the drug sponsor, Brainstorm Cell Therapeutics, announced that it had filed its application for FDA approval, called a Biologics License Application (BLA).
Mark your calendars. On September 27th, the FDA will hold an Advisory Committee meeting (AdCom) to discuss the BLA. December 8th is the deadline (PDUFA date) for the FDA’s biologics division (CBER) to make its decision about NurOwn’s approval.
Last month, FDA CBER’s Cellular, Tissue, and Gene Therapies Advisory Committee published the docket where people can submit public comments about the NurOwn application. The NurOwn (Debamestrocel) docket number is FDA-2023-N-2608.
At the AdCom, both the FDA and Brainstorm will present their views of the evidence to an independent panel of experts. At the end of that day, those AdCom members will make a recommendation to the FDA about the therapy’s approval. Historically, the FDA has followed the AdCom recommendations, but it is not bound to do so.
At the Biopharma Congress earlier this year, the new FDA Commissioner, Dr. Robert Califf spoke about the advisory committee process:
“The purpose of these meetings is to provoke discussion and debate, and not always agree…. The purpose is to get advice, and the best advice is not whether this drug should be approved. That decision should be made by full time civil servants.”
At the AdCom, people in the ALS and scientific community will get to hear from the trial’s principal investigators about their view of the evidence and about their clinical observations of patients who participated in the NurOwn trial and Expanded Access Program.
If you want to learn more about the data regarding NurOwn, you can read some of the peer-reviewed publications and scientific presentations. In the Phase 2 NurOwn trial in 2015 (NCT02017912), 36 of 48 trial participants received one dose of NurOwn. Those results were published in Neurology. In the Phase 3 trial (NCT03280056) that ran from 2017-2020, 95 of 189 received three doses of NurOwn, once every two months. You can read the Phase 3 trial data, erratum and supplement published in Muscle & Nerve.
Brainstorm has presented the biomarker data at several biotech conferences: first with Dr. Robert Brown at the ALS-MND Conference and most recently with Brainstorm’s CEO and biostatistician Dr. Stacy Lindborg, PhD at the Gordon Research Conference.
In 2021 and again in 2022, a small group of Phase 3 trial participants received an additional six doses of NurOwn in an Expanded Access protocol (EAP). The results of the EAP and the biomarker work have not yet been published.
A few days before the FDA’s September 27th Advisory Committee meeting, Brainstorm and the FDA will file their respective Briefing Documents explaining their view of the evidence regarding NurOwn’s approval. The docket will also identify who the members of the Advisory Committee will be. You can find that information here.
If you want to familiarize yourself with how an AdCom is conducted, you can listen to the recent AdCom meetings with the CDER division of the FDA discussing the approval of AMX0035 (Relyrio) and Tofersen (Qalsody); or CBER discussing Sarepta’s therapy for Duchenne Muscular Dystrophy. These recordings are relevant as two involved the Accelerated Approval pathway and all involved the FDA exercising regulatory flexibility to approve those drugs.
Given that currently enrolled trials for sporadic ALS have estimated completion dates extending into 2025 and 2026, it is likely that NurOwn will be the only sporadic ALS therapy that will be submitted for FDA approval for a few years.
We hope this summary provides some insight into some of the promising ALS therapies in clinical trials right now for sporadic ALS. Next quarter, Synapticure on Science will be discussing investigational therapies for people with genetic mutations causing ALS as well some important observational trials. If you want to have a conversation about any of the investigational therapies mentioned in this science summary, or any other clinical trials, please reach out to our care coordinators to schedule a telehealth appointment.
Author: Neuromuscular Specialist Dr. Danielle Geraldi-Samara
Synapticure’s ALS care team is led by Dr. Danielle Geraldi-Samara. For over 15 years, Dr. Geraldi-Samara has focused on diagnosing and treating ALS patients, beginning with her neuromuscular fellowship at the Mount Sinai School of Medicine. Dr. Geraldi-Samara has long-recognized the need for her patients to easily access care at an ALS clinic in their community; and thus, she established an ALS/Neuromuscular clinic from the ground up at NYU-Brooklyn. She also served as a supervising Neuromuscular Specialist at Northwell Health’s multidisciplinary ALS clinic in New York.
To read the amazing reviews about Dr. Samara’ and the rest of Synapticure’s neurologists, click here.
1 In an upcoming Synapticure on Science, we will be reviewing ALS drug trials that target genetic forms of ALS. In that volume, we will spend more time detailing the science underlying polyQ repeats in the ATXN2 gene.
2 In post-hoc analyses, rapidly declining participants on pridopidine with definite or probable ALS who were early in the disease (less than 18 months from symptom onset) had substantially less decline in the ALSFRS-R total score: -7.51 points on Pridopidine compared to -12.71 points on placebo (unadjusted p-value=0.04).
3 If you want to learn more about NfL and how it impacted the approval of Tofersen, you can watch this May 2nd NEALS webinar with Dr. Timothy Miller of Washington University, the trial’s principal investigator.
4 Comparatively, over 28 weeks in the overall intent-to-treat population (n=108), Tofersen- treated participants experienced a 55% reduction in plasma NfL compared to a 12% increase in placebo-treated participants (difference in geometric mean ratios for Tofersen to placebo: 60%; nominal p<0.0001).
5 Clinically, AP would be categorized as minimal, mild, moderate, severe articulatory imprecision and/or blending of phoneme production. As outlined in this study, “AP is a measure of the match between the expected and observed acoustic features for each phoneme. The algorithm assesses how well the acoustics of each phoneme correspond to the acoustics of the expected phoneme in spoken English…. For ease of interpretation, articulatory precision was projected onto a 0–10 scale (higher scores are indicative of more precise articulation)."
6 The gender, racial and ethnic distribution in the Pridopidine trial is as follows: 35% female; 7% Hispanic; 6% African American and 4% Asian. There were no Native Americans enrolled.
7 We will also bring you the trial data from their concurrent Pridopidine trial in Huntington’s Disease. That data is scheduled to be presented in more detail at an upcoming scientific conference.
8 The CNM-Au8 trial enrolled 161 participants. The gender, racial and ethnic distribution is as follows: 38% female; 4% Hispanic; 1% African American. There were no Asians or Native Americans enrolled.
Your first consultation with a care navigator is at no cost. Give us a call at 855-255-5917 for details.
